Getting Started
The WebMO editor is a Java applet that facilitates the entry of the structure of interest in a three dimensional environment. The editor has three basic tools:- View Tool - Allows the rotation, translation, and zooming of the molecule
- Build Tool - Allows the addition of new atoms, bonds, and fragments
- Adjust Tool - Allows the changing of bond distances, angles, and dihedral angles, as well as the translation, rotation, and duplication of fragments
In addition, the following menus provide access to the remaining functionality of the WebMO editor:
Tutorial
If you prefer to learn by example, the following provide a demonstration of the WebMO editor.File Menu
New
This menu item brings up a blank editor window, erasing any molecule that may currently be draw.Preferences
The preferences menu item brings up a preferences dialog, allowing display and 'undo' preferences to be set.Close
The close menu item closes the editor dialog and returns you to the "Build Molecule" page.Edit Menu
Undo
This menu item undoes the last change made to the molecule.Redo
This menu item reverts the molecule to the form prior to the last undo.Find
This menu item translates the molecule to the center of the editor window, and set the origin of rotation to the center of the molecule. It is useful if the molecule has inadvertently been translated off the screen and is "lost".Tools Menu
The tools menu contains menu items which select the current editor tool. The three basic tools contained in the editor are:- View Tool - Allows the rotation, translation, and zooming of the molecule
- Build Tool - Allows the addition of new atoms, bonds, and fragments
- Adjust Tool - Allows the changing of bond distances, angles, and dihedral angles, as well as the translation, rotation, and duplication of fragments
- Z-Matrix Editor Tool - Allows advanced control over the z-matrix description of the current molecule
View Tool
View mode can be accessed through the Tools:View menu option, or by clicking on the translate or zoom
or zoom buttons on the tool bar. When in view mode, the View
menu provides access to the translate, rotate, and zoom options. The options will allow the manipulation
of the molecule in 3-D space.
buttons on the tool bar. When in view mode, the View
menu provides access to the translate, rotate, and zoom options. The options will allow the manipulation
of the molecule in 3-D space.
Rotate
After selecting the rotate option through the View:Rotate menu option, you are free to rotate the molecule. Dragging the mouse will rotate the structure in the about the x and y axes (in the plane of the screen), while ctrl-drag will rotate the molecule about the z axis (perpendicular to the screen).Translate
After selecting the translate option through the View:Translate menu option, you are free to translate the molecule. Dragging the mouse will translate the structure in the x-y plane.Zoom
After selecting the rotate option through the View:Zoom menu option, you are free to zoom the molecule. Dragging the mouse upward will increase the size of the molecule, while dragging downward will decrease its size.Build Tool
Build mode can be accessed through the Tools:Build menu option, or by clicking the button resembling a molecule on the tool bar. In build mode, atoms and bonds are added
to the existing structure.
on the tool bar. In build mode, atoms and bonds are added
to the existing structure.
Adding an atom
To add an atom, simply single click on the editor window. To add both an atom and a bond, click and drag from an existing atom to a blank space on the editor window.Adding a bond
To add a bond, simply click and drag between two existing atoms. To create a double or triple bond, click and drag repeatedly until the appropriate bond order is achieved.Changing atoms
If you wish to add a carbon, hydrogen, oxygen, or nitrogen atom, simply select the appropriate atom from the Build menu. Otherwise, choose Build:Other or the tool bar button to bring up a periodic table from which any atom can be selected.
tool bar button to bring up a periodic table from which any atom can be selected.
Fragments
The Fragments menu option allows the insertion of pre-created fragments into the editor. Upon selecting this option, a dialog will appear listing the available fragments. Choose the desired fragment, and select OK to close the dialog. Click to add the selected fragment. The fragment can then be translated and rotated as desired by first selecting the fragment, and then using the Adjust:Translate/Rotate Selection menu option.Adjust Tool
Adjust mode can be accessed through the Tools:Adjust menu option, or by clicking the cursor button
on the tool bar. Adjust mode allows the alteration of bond distances, angles, and dihedral angles,
as well as selecting and deleting atoms. Atoms and bonds are selected by clicking on the center of the
atom or bond that you wish to select. Multiple atoms can be selected by shift-clicking. Shift-clicking an
unselected object adds it to the selection set. Shift-clicking a selected object removes it from the selection
set. An entire area can be selected by clicking a dragging a box around a group of atoms and bonds. Clicking
in the background will select the entire molecule. Ctrl-clicking an atom will bring up a hybridization context
menu, while Ctrl-clicking a bond will bring up a bond order context menu.
button
on the tool bar. Adjust mode allows the alteration of bond distances, angles, and dihedral angles,
as well as selecting and deleting atoms. Atoms and bonds are selected by clicking on the center of the
atom or bond that you wish to select. Multiple atoms can be selected by shift-clicking. Shift-clicking an
unselected object adds it to the selection set. Shift-clicking a selected object removes it from the selection
set. An entire area can be selected by clicking a dragging a box around a group of atoms and bonds. Clicking
in the background will select the entire molecule. Ctrl-clicking an atom will bring up a hybridization context
menu, while Ctrl-clicking a bond will bring up a bond order context menu.
Bond length
This menu item is available only when exactly two atoms are selected. This menu item brings up a dialog which will allow the bond distance between two atoms to be altered. All distances are displayed in angstroms. This option is also accessible through the tool bar .
.
Bond angle
This menu item is available only when exactly three atoms are selected. This menu item brings up a dialog which will allow the bond angle between three atoms to be altered. All bond distances remain unchanged. This option is also accessible through the tool bar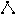 .
.
Dihedral angle
This menu item is available only when exactly four atoms are selected. This menu item brings up a dialog which will allow the dihedral angle between four atoms to be altered. All bond distances and bond angles remain unchanged. This option is also accessible through the tool bar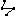 .
.
Select molecule
This menu item will completely select any molecule which is current partially selected. For example, to select a large fragment, simply click one atom and then choose Adjust:Select Molecule.Rotate selection
This menu item allows the selected fragment to be freely rotated. Dragging the mouse will rotate the fragment in the about the x and y axes (in the plane of the screen), while the a Ctrl-drag will rotate the fragment about the z axis (perpendicular to the screen).Translate selection
This menu item allows the selected fragment to be freely translated. Dragging the mouse will translate the fragment in the x-y plane, while a Ctrl-drag will translate the fragment in the z-direction.Find selection
This option will center the editor on the selected atoms and bonds, and adjust the zoom factor to an appropriate level.Duplicate selection
This option allows a copy of the selected portion of the molecule to be inserted into the editor. Select the desired portion of the molecule, and choose Adjust:Duplicate Selection. Click to add the fragment. The fragment can then be translated and rotated as desired by first selecting the fragment, and then using the Adjust:Translate/Rotate Selection menu option.Delete selection
This option will delete the selected atoms and bonds from the molecule.Invert chiral center
This menu item is available only when exactly one atom is selected. This menu item will attempt to invert a chiral center be switching the location of two substituent of the chiral center.Z-Matrix Editor
The z-matrix editor allows complete control over the z-matrix for current molecule. To use the z-matrix editor, use the following procedure. First, reorder the atoms in the z-matrix if necessary. Note that atoms can only reference atoms ABOVE them in the z-matrix. To reorder the atoms, simply change the numbering on the far left hand side (decimals OK) and click the "ReOrder" button; the atoms will be resorted in numerical order. Second, change the connectivity definitions in the z-matrix (NA, NB, NC). Change the number(s) in the appropriate column and click the "ReConnect" button. Finally, mark coordinates to be optimized, fixed, or scanned as appropriate using the drop-down box next to the coordinate. Scan information is specified at the bottom of the editor using the start and stop values, as well as the desired number of steps.Clean-Up
Add hydrogens
This menu item will fill the unfilled valencies of all the atoms in the molecule with hydrogen atoms.Hybridization
This menu item will attempt to assign the correct hybridization to every atom in the molecule.Geometry
The menu item will use the assigned hybridization in an attempt to generate a starting geometry for the molecule that is somewhat close to the optimized geometry.Comprehensive
This menu item will attempt to fill empty valencies with hydrogens, assign the correct hybridization, and then optimize the starting geometry in a single step. This option is also accessible from the tool bar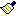 .
.
Selection Only
Activating this menu item will cause future clean-up operations to be restricted to the currently selected atoms. All other portions of the molecule will be left unaffected.Help Menu
Index
This menu item brings up the help file you are reading now.
| Program Help | Editor Help | Administration Help |